


What is Protein?
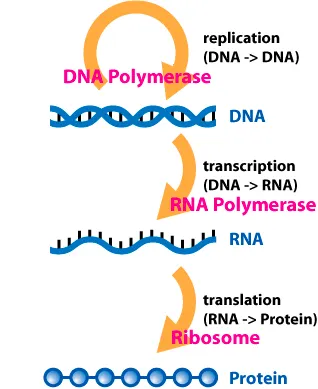
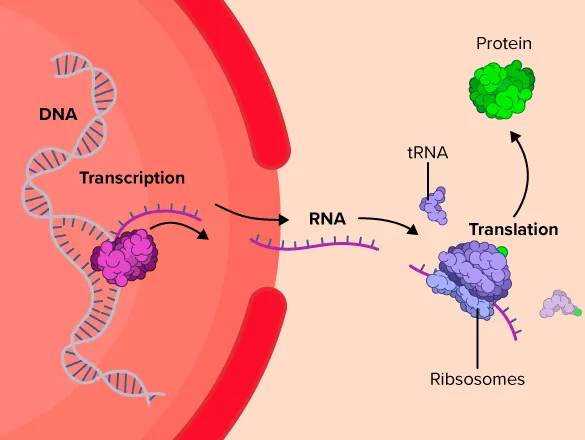
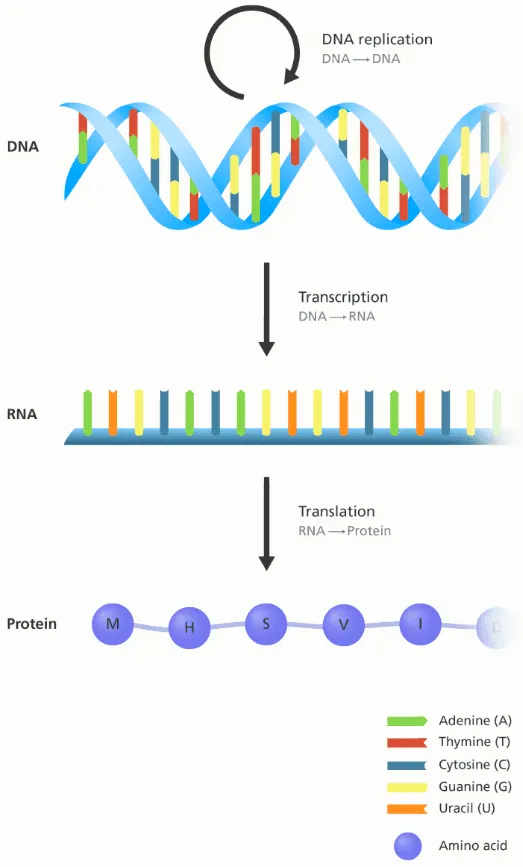
Central Dogma: DNA → RNA → Protein
Source: Wikipedia
Source: CK-12 Foundation
The central dogma is the most important concept in modern molecular biology. Below is the explanation from Wikipedia:
The central dogma of molecular biology is an explanation of the flow of genetic information within a biological system. It is often stated as "DNA makes RNA, and RNA makes protein", although this is not its original meaning. It was first stated by Francis Crick in 1957, then published in 1958:
Source: Genome Research Limited
The Central Dogma. This states that once "information" has passed into protein it cannot get out again. In more detail, the transfer of information from nucleic acid to nucleic acid, or from nucleic acid to protein may be possible, but transfer from protein to protein, or from protein to nucleic acid is impossible. Information here means the precise determination of sequence, either of bases in the nucleic acid or of amino acid residues in the protein.
There are three well-known information-carrying biopolymers: DNA, RNA, Protein
The smallest “tokens” of each biopolymers are:
•
DNA: A, T, G, C
•
RNA: A, U, G, C
•
Protein: 20 amino acids
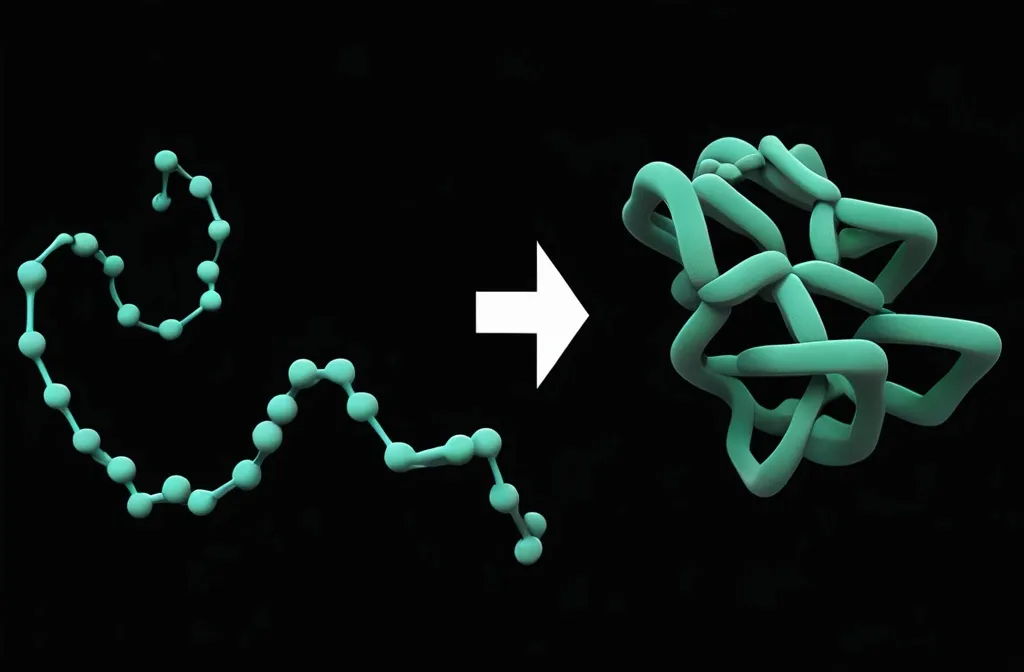
What is Protein Folding?
From 1D to 3D
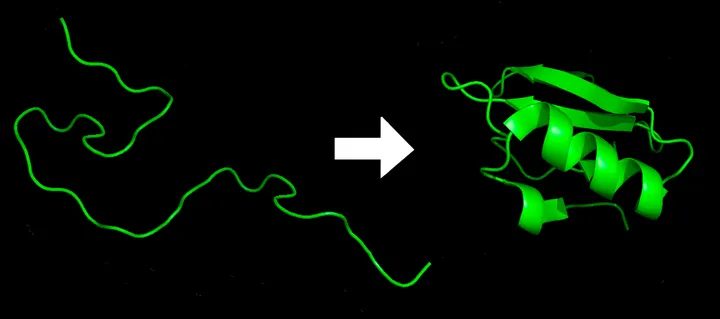
Source: Wikipedia
Protein folding is literally folding a (relatively) unstable 1D string into a (relatively) stable 3D structure.
•
The 1D string is a linear chain of amino acids.
•
Proteins need to be folded to become biologically functional. In other words, the structure defines its function!
•
The folded 3D structure is not fixed. Rather, it can change if the local environment (temperature, pH, solvent, interacting biomolecules, …) changes.
Protein structure hierarchy
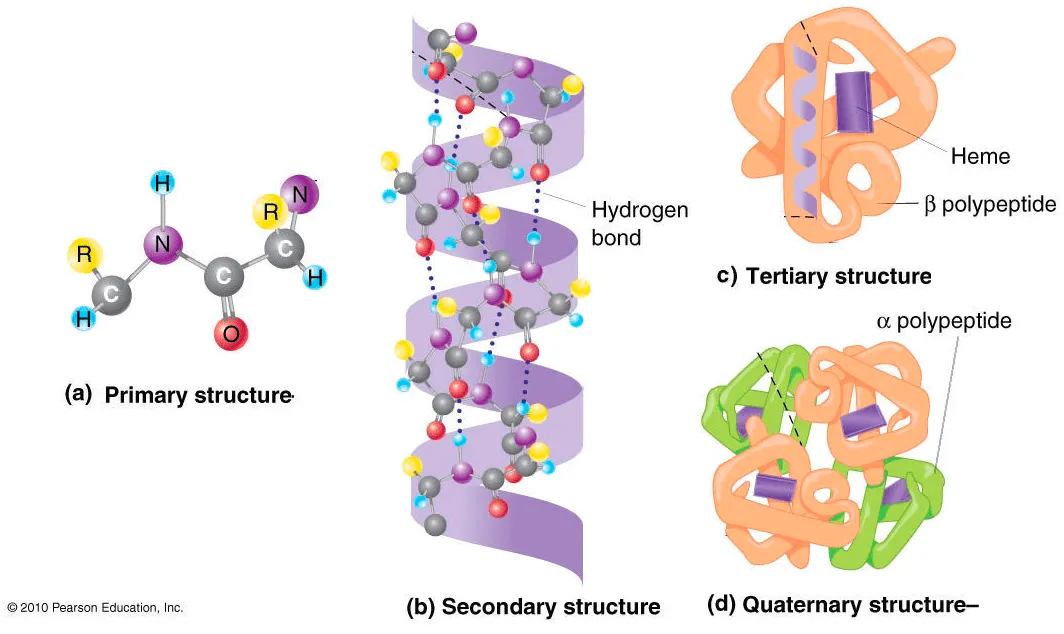
Source: iGenetics 3rd ed.
We often categorize the hierarchy into four levels.
1.
Primary structure: Linear amino acid sequence (synthesized by ribosome)
2.
Secondary structure: Typical local structure patterns. Primary structure first builds various secondary structure since this is easily stabilized by intramolecular hydrogen bonds.
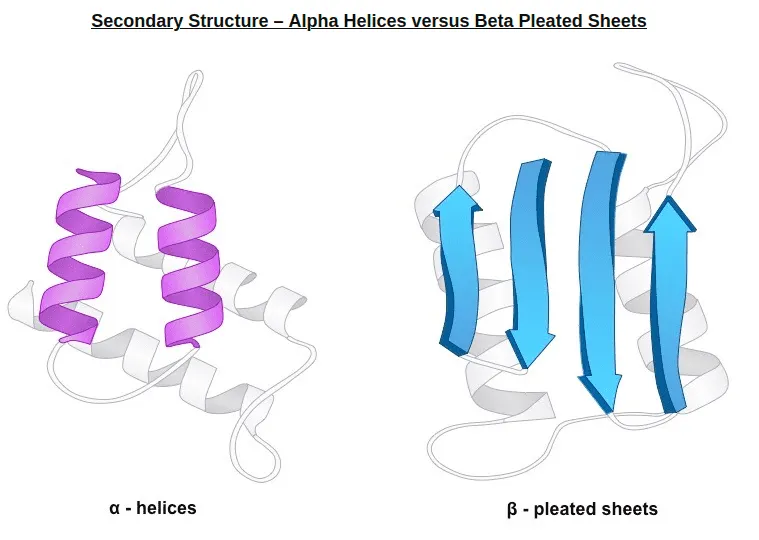
From bioninja.com. Two popular secondary structure: alpha helix, beta sheet
3.
Tertiary structure: Folded structure of single polypeptide chain. Usually, hydrophilic sides are facing the aqueous environment surrounding the protein, and the hydrophobic sides are facing the hydrophobic core of the protein.
4.
Quaternary structure: Assembly of tertiary structures. i.e. multi-chain structure.
Structure defines Function
Traditionally (and still), protein structure is determined by various methods:
•
X-ray crystallography
•
Nuclear Magnetic Resonance (NMR)
•
Cryo-EM
•
…
Each method has its own pros and cons, but is used to build ground truth structure labels.
However, structure determination requires extensive trial & error, years of work (almost the entire PhD  ), and expensive equipments.
), and expensive equipments.
), and expensive equipments.c.f. commonly observed structural motifs
Modeling of protein folding
The search space of protein folding is enormous. Brute-force approach () is not even possible to see the results. But in biological systems (e.g. our body), it is done in a few milliseconds ~ few hours, which is fascinating.
Several computational methods have been developed to predict the structure.
•
Molecular dynamics (MD) simulations
•
Anton: massively parallel supercomputer
•
Rosetta
•
…
But these tools comes with high computational cost, which makes them applicable to only short polypeptides.
CASP: Critical Assessment of protein Structure Prediction
Since the late 1960s, understanding and simulating the protein folding process has been an important challenge for computational biology.
In 1994, a biennial competition called CASP has begun to facilitate the development of computational tools to predict folded structure of proteins.