



Introduction
PDB data를 다루기 위해 기본적으로 알아야 하는 Python, Pandas, Numpy, Scipy, Matplotlib, biopython tutorial이다.
Introduction 부분에서는 Python 기초를 다룬다.
•
Jupyter Notebook
•
Variable 할당, data type
•
Slicing
•
For loop
•
Logic Statement
File Parsing
•
os library
◦
os.getcwd()
◦
os.listdir()
◦
os.path.join()
•
File reading
with open(<filepath:str>, "r") as f:
data = f.readlines()
Python
복사
•
Find patterns with for loop & logic statements
•
Regular Expressions
Processing Multiple Files and Writing Files
•
os library + glob library
import os
file_loc = os.path.join('data', 'PDB_files', '*.pdb')
import glob
filenames = glob.glob(file_loc)
Python
복사
•
File writing
with open(<filename:str>, "w") as f:
f.write(<stringtowrite:str>)
Python
복사
r | read-only | pointer at the beginning |
r+ | read & write | pointer at the beginning |
w | write-only | if not exist → truncates file or creates
pointer at the beginning |
w+ | read & write | if not exist → truncates file or creates
pointer at the beginning |
a | append | if not exist → create file
pointer at the end |
a+ | read & append | if not exist → create file
pointer at the end |
•
String formatting
Working with Pandas
•
pandas library
◦
pd.read_csv()
◦
df.loc[<row idx>, <col name>]
◦
df.iloc[<row idx>, <col idx>]
◦
df.head()
◦
df.tail()
◦
df.sort_values()
Linear Regression
•
scipy.stats module
◦
stats.linregress(x, y)
•
Conditional retrieval from dataframe
df.loc[<condition>, <col name>]
df[<condition>]
Creating Plots in Jupyter Notebooks
•
matplotlib.pyplot module
◦
plt.scatter(x, y)
◦
plt.plot(x, y)
◦
plt.xlabel(<str>)
◦
plt.ylabel(<str>)
◦
plt.annotate(<str>, xy=(<xloc:float>, <yloc:float>)
◦
plt.savefig(<str>, dpi=int)
•
seaborn library
◦
sns.regplot(x, y)
Nonlinear Regression
•
scipy.optimize module
◦
curve_fit(function, x, y)
Analyzing MMCIF files using biopython
Objectives
•
Biopython library에 대한 이해
•
Biopython으로 MMCIF file을 다루는 방법
Reading MMCIF Files
mmCIF (PDBx/mmCIF라고도 불린다) 파일을 biopython library로 다루는 실습이다.
매우 유용한 실습이니 잘 공부해 둘 것!
mmCIF 파일은 category, attribute으로 나열, 구성되어 있다.
_citation.year
Plain Text
복사
Biopython
Biopython simplifies tasks such as working with DNA and protein sequences, interacting with online databases like GenBank and UniProt, and reading molecular format files like mmCIF and PDB.
Bio.PDB → PDB 관련 resource
MMCIF2Dict function
•
.cif 파일을 dictionary 형태로 읽어낼 수 있는 함수이다.
from Bio.PDB.MMCIF2Dict import MMCIF2Dict
pdb_info = MMCIF2Dict('data/PDB_files/1mbn.cif')
Python
복사
MMCIFParser function
•
.cif 파일을 Structure type으로 읽어낼 수 있는 함수이다.
from Bio.PDB.MMCIFParser import MMCIFParser
parser = MMCIFParser(QUIET=True)
structure = parser.get_structure('myoglobin', 'data/PDB_files/1mbn.cif')
Python
복사
SMCRA: Structure > Model > Chain > Residue > Atom
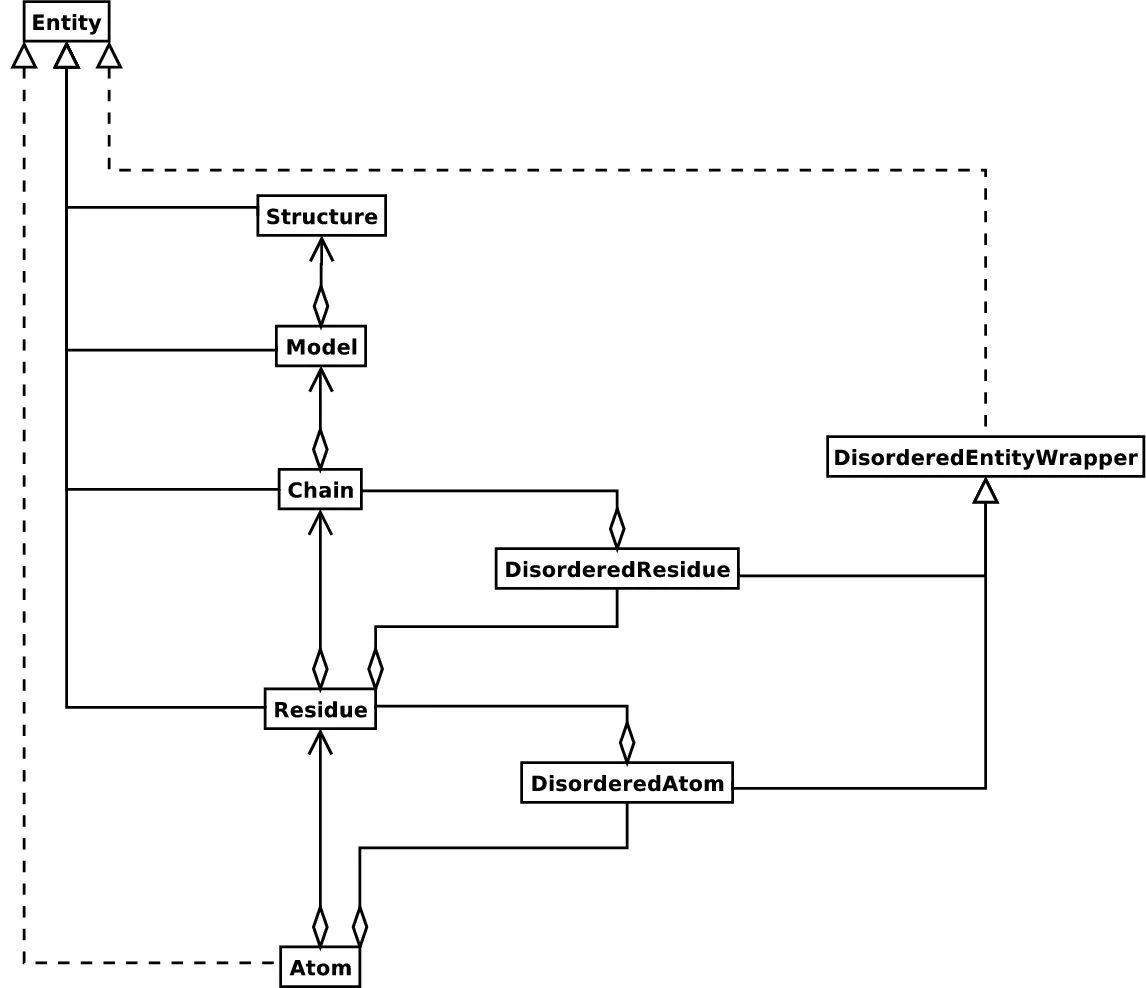
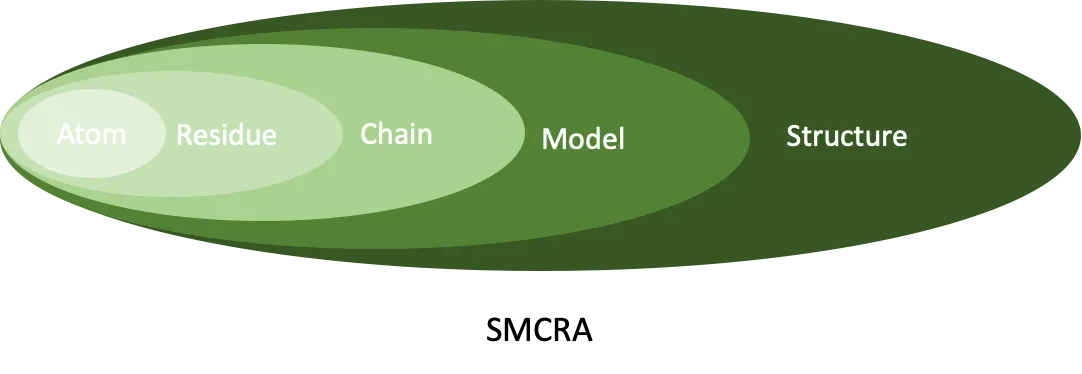
Structure object는 molecule을 hierarchical 하게 표현한다.
•
Structure = Model로 구성
•
Model = Chain으로 구성
•
Chain = Residue로 구성
•
Residue = Atom으로 구성
Measuring 3D properties
하나의 Structure data로부터 atom들을 얻고, 특정 atom 주변에 있는 residue를 찾아내는 것을 다룬다.
MolSSI 페이지에 있는 1mbn.cif 파일로 하는 실습을 먼저 수행해보자.
아래 풀이는 1a1t.cif 파일을 분석하는 코드이다.
from Bio.PDB.MMCIFParser import MMCIFParser
from Bio.PDB import NeighborSearch
parser = MMCIFParser(QUIET=True)
structures = parser.get_structure('1a1t', 'data/PDB_files/1a1t.cif')
print(len(structures)) # 25 -> list of 25 models
structure = structures[0] # select only one model
atoms = structure.get_atoms()
atom_list = list(atoms)
zincs = [] # multiple zincs exists
for atom in atom_list:
if atom.element == 'ZN':
zincs.append(atom)
zinc1, zinc2 = zincs[0], zincs[1]
def find_neighbor(atom_list, ref_atom, cutoff_dist):
nei_search = NeighborSearch(atom_list)
neighbors = nei_search.search(ref_atom.get_coord(), cutoff_dist)
ref_residue = ref_atom.get_parent()
for negi in neighbors:
residue = nei.get_parent()
if residue != ref_residue:
print(nei.element, residue.get_resname(), residue.get_id()[1])
find_neighbor(atom_list, zinc1, 4)
find_neighbor(atom_list, zinc2, 4)
Python
복사
Retrieving Information from the PDB using the Web API
Objectives
•
REST API에 대한 이해
•
REST API와 Python을 활용해서 data를 가져오는 방법
REST: Representational State Transfer
API: Application Programming Interface
서로 다른 software system끼리 소통하기 위한 도구.
File Download using Biopython
PDB에서 structure file을 다운로드하는 방법
1.
PDBList() instance를 만든다.
2.
pdb_list.retrieve_pdb_file(pdb_id, pdir=<dirname:str>, file_format="mmCif") 함수를 활용하여 파일을 저장한다.
PDB ID, 저장할 directory, file format 등을 parameter로 넣어준다.
PDB Data API
Web API를 사용해 structure file말고 PDB entry 자체에 대한 정보를 얻을 수 있다.
아래와 같은 format으로 사용한다.
https://data.rcsb.org/rest/v1/core/<TYPE_OF_RESOURCE>/<IDENTIFIER>
Python
복사
예를 들어, 4hhb의 full entry를 얻으려면,
https://data.rcsb.org/rest/v1/core/entry/4hhb
Python
복사
Programmatic Access of APIs
URL을 직접 활용하여 다운로드 해보자.
import requests
data = requests.get("https://data.rcsb.org/rest/v1/core/entry/4hhb")
data.status_code # 200 if successful, 404 if not found
info_4hhb = data.json() # json format으로 load
Python
복사
PDB Search API
PDB는 Search API도 제공한다. 어떤 keyword, host species, sequence similarity 등 다양한 기준으로 검색할 수 있다. 이 API를 사용하는 방법은 꽤 복잡하지만, 다음 링크에 잘 정리되어 있다.
Search하기 위한 URL format은 다음과 같다.
https://search.rcsb.org/rcsbsearch/v2/query?json={search-request}
Plain Text
복사
import json
my_query = {
"query": {
"type": "terminal",
"service": "full_text",
"parameters": {
"value": '"oxygen storage"'
}
},
"return_type": "entry"
}
my_query = json.dumps(my_query)
data = requests.get(f"https://search.rcsb.org/rcsbsearch/v2/query?json={my_query}")
results = data.json()
results
Python
복사
따로 request에 명시하지 않으면 최대 10개까지의 결과만 제공한다. 그래도 total_count 에 실제 검색되는 개수를 보여주니, 이를 참고해서 request 하자.
Using Biopython to Analyze Search Results
1.
PDB Search API를 활용해 원하는 조건에 해당하는 단백질의 pdb id list 얻음.
2.
Biopython의 PDBList 를 활용해 structure file 다운로드.
3.
각 단백질 구조 분석
예시: 어떤 atom과 특정 threshold distance 안에 존재하는 atom들의 residue count
Molecular Visualization with iCN3D
TBU