


Introduction
If you need some background information about protein folding, refer to  Introduction to Protein Folding
Introduction to Protein Folding 
Introduction to Protein Folding AlphaFold Multimer (AF2-Multimer)
Multimeric structure prediction model
Major differences compared to vanilla AF2
1.
Multi-chain featurization
Since AF2-Multimer deals with more than one chain, authors used three features to represent multimeric state.
•
asym_id: unique integer per chain
•
entity_id: unique integer for each set of identical chains
•
sym_id: unique integer within a set of identical chains
example: A3B2 stoichiometry
2.
Multi-chain cropping
Since the number of residues that can be handled is limited (by memory & computation), authors applied two strategies to crop the structure.
•
Per-chain contiguous cropping (in sequence space)
The chains are first randomly shuffled and then some contiguous regions are selected per chain until the total number of residues reach budget.
This cropping ensures more than certain amount of each chain to be included.
•
Inter-chain spatial cropping (interface-biased, structure space)
This cropping targets interface regions, which are the spatially nearest neighbors, as defined by distances between C coordinates.
3.
Symmetry handling
•
Greedy heuristic approach to deal with multi-chain permutation alignment
When computing the losses for homomeric components, permutation symmetry becomes important. Since the order of the predicted chains and the ground-truth chains may differ between chains with identical sequences, authors used greedy approach to find good chain mappings.
4.
Loss
•
FAPE loss cutoff
◦
intra-chain: 10Å (same as vanilla AF2)
◦
inter-chain: 30Å (new in AF2-Multimer)
•
(new) chain center-of-mass loss term
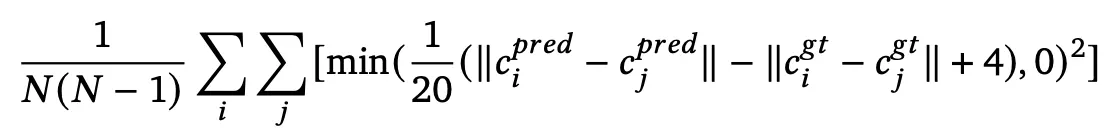
push apart different chains (clamped if the error is -4Å or greater)
Goal: to prevent the model from predicting overlapping chains
•
(modified) clash loss
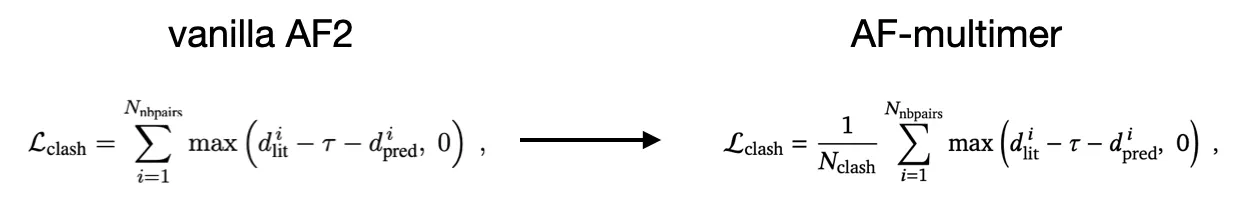
average, rather than sum
Goal: stabilize the loss (since there maybe many clashes if Ncycle is small - due to black hole initialization)
5.
Architecture
•
Template stack
Swapped the order of attention and triangular multiplicative update layers
Changed the aggregation of template embeddings
•
Evoformer
Moved the outer product mean to the start of the Evoformer block
Results
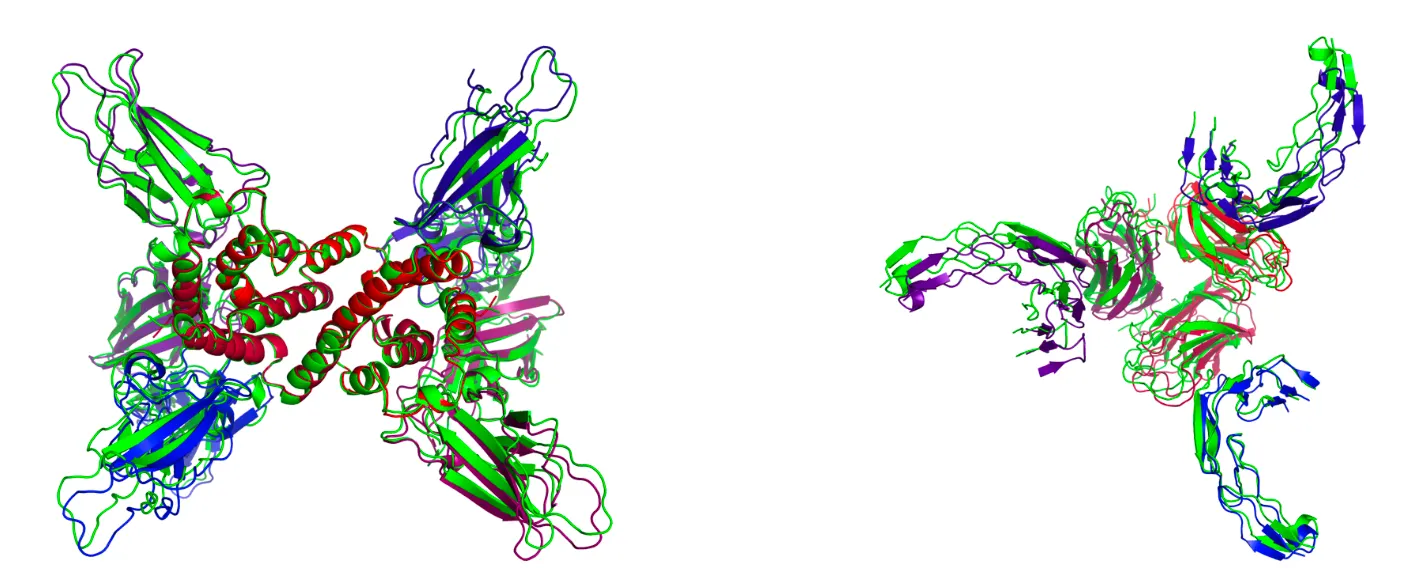
Structures
Mean DockQ score
Confidence score (interface pTM) vs. DockQ score
AF-multimer predicts better individual chain than AF2?
Discussion
•
Multi-chain version of AF2, with some modifications